
Ορισμένες ασθένειες, παρότι κάνουν την εμφάνισή τους αρκετά σπάνια στον ανθρώπινο πληθυσμό, δεν παύουν να προβληματίζουν την επιστημονική κοινότητα και να αποτελούν συνεχή πρόκληση για τους ερευνητές. Πολλές από αυτές, ωστόσο, είναι άγνωστες στο ευρύ κοινό παρά το εξαιρετικό ενδιαφέρον που παρουσιάζουν και τα εντυπωσιακά άλματα που, με την πάροδο του χρόνου, έχει κάνει η επιστήμη με σκοπό την καλύτερη αντιμετώπισή τους.
Η νόσος Pompe (ή αλλιώς: γλυκογονίαση τύπου ΙΙ, ανεπάρκεια της όξινης μαλτάσης) αποτελεί μεταβολικό μονογονιδιακό νόσημα το οποίο κληρονομείται με αυτοσωμικό υπολειπόμενο τρόπο και οφείλεται στην πλήρη ή μερική έλλειψη της α-γλυκοσιδάσης (ή όξινης μαλτάσης), ενός ενζύμου το οποίο είναι υπεύθυνο για την διάσπαση του γλυκογόνου σε γλυκόζη. Ως αποτέλεσμα, προκαλείται συσσώρευση του γλυκογόνου στα λυσοσώματα των σωματικών κυττάρων.
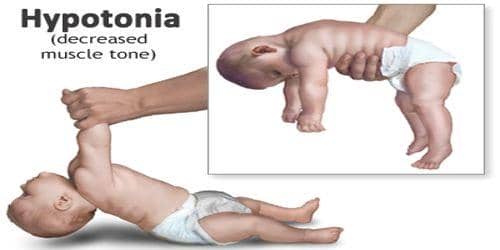
Η ασθένεια αυτή χαρακτηρίζεται από ευρύ φάσμα συμπτωμάτων και διακρίνεται, με βάση την βαρύτητα αυτών και την ηλικία έναρξής τους, σε δύο μορφές: την κλασική βρεφική μορφή (Infantile-onset Pompe disease – IOPD) και την όψιμη μορφή (Late-onset Pompe disease – LOPD). Η πρώτη, εκδηλώνεται συνήθως εντός των πρώτων τεσσάρων μηνών της ζωής με γενικευμένη μυϊκή αδυναμία (μυοπάθεια), υποτονία, αναπνευστικά προβλήματα, δυσκολία σίτισης, αδυναμία ανάπτυξης και καρδιομεγαλία που οδηγεί σε καρδιακή ανεπάρκεια. Χωρίς αντιμετώπιση, αυτή η μορφή της ασθένειας οδηγεί σε θάνατο εντός των δύο πρώτων χρόνων ζωής. Η όψιμη μορφή χαρακτηρίζεται από προοδευτική μυϊκή αδυναμία και αναπνευστική ανεπάρκεια, ενώ σπανίως επηρεάζεται η καρδιά. Διακρίνεται στην μη κλασική βρεφική μορφή η οποία εκδηλώνεται εντός του πρώτου έτους ζωής και στην όψιμη μορφή που εκδηλώνεται μετά το πρώτο έτος ζωής. Όσον αφορά την όψιμη μορφή της νόσου, αποτελεί τον ηπιότερο τύπο καθώς χαρακτηρίζεται από μερική έλλειψη της α γλυκοσιδάσης.
Η νόσος Pompe οφείλεται σε μεταλλάξεις στο γονίδιο GAA, το οποίο βρίσκεται στην θέση 17q25.3 του χρωμοσώματος 17. Το γονίδιο αυτό κωδικοποιεί το ένζυμο α-γλυκοσιδάση/όξινη μαλτάση (alpha-glucosidase/acid maltase) το οποίο προκαλεί την διάσπαση του γλυκογόνου σε γλυκόζη, που αποτελεί βασική πηγή ενέργειας των κυττάρων. Η διαδικασία αυτή λαμβάνει χώρα στα λυσοσώματα των σωματικών κυττάρων. Μεταλλάξεις στο γονίδιο GAA έχουν ως αποτέλεσμα την συσσώρευση μεγάλης ποσότητας γλυκογόνου στα λυσοσώματα, κυρίως των μυοκαρδιακών κυττάρων και των σκελετικών μυϊκών ινών, το οποίο είναι τοξικό για τα ίδια και έχει ως αποτέλεσμα την καταστροφή οργάνων και ιστών και την εμφάνιση των συμπτωμάτων της νόσου Pompe.

Η βρεφική μορφή της ασθένειας εμφανίζεται µε συχνότητα 1:138.000, ενώ η όψιμη μορφή της µε συχνότητα 1:57.000. Μετά από μελέτη 7 μεταλλάξεων που αντιπροσωπεύουν το 29% του συνόλου και με βάση το ισοζύγιο Hardy – Weinberg, εκτιμήθηκε ότι η νόσος Pompe εμφανίζεται με συχνότητα 1:40.000. Η συχνότητα για την Ευρώπη είναι 1:100.000 (βρεφική μορφή) και 1:60.000 (όψιμη μορφή), ενώ την Ελλάδα έχουν καταγραφεί μόνο 7 ασθενείς με τη νόσο Pompe.
Όσον αφορά τις θεραπευτικές προσεγγίσεις, η γονιδιακή θεραπεία της νόσου Pompe βρίσκεται σε πειραματικό στάδιο. Προς το παρόν, μελετάται ο τρόπος με τον οποίο θα πραγματοποιηθεί εισαγωγή του φυσιολογικού γονιδίου GAA στο γονιδίωμα των ασθενών, έτσι ώστε να γίνει δυνατή η παραγωγή της απαιτούμενης ποσότητας της α γλυκοσιδάσης. Ωστόσο, αν η διάγνωση της ασθένειας γίνει σε αρχικά στάδια, συχνά εφαρμόζεται θεραπεία ενζυμικής υποκατάστασης κατά την οποία χορηγείται στους ασθενείς το ανθρώπινο ανασυνδυασμένο ένζυμο, μέθοδος η οποία κάποιες φορές έρχεται αντιμέτωπη με το ανοσοποιητικό τους σύστημα, γι’ αυτό και χρειάζεται περαιτέρω μελέτη. Εκτός αυτών, συχνά εφαρμόζονται μέθοδοι αντιμετώπισης των κλινικών συμπτωμάτων όπως λήψη φαρμακευτικής αγωγής, φυσιοθεραπεία, λογοθεραπεία και τραχειοστομία (σε περιπτώσεις αναπνευστικής ανεπάρκειας ή μακρογλωσσίας).
Πηγές:
[1]. https://www.omim.org/entry/232300
[2]. https://www.omim.org/entry/606800
[3]. https://www.ncbi.nlm.nih.gov/books/NBK1261/
[4]. https://www.ncbi.nlm.nih.gov/pubmed/16838077
[5]. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC1149727/pdf/biochemj00170-0210.pdf

