Πολιτισμός
 Τελευταίο άρθροΤο Evia Film Project επιστρέφειSpecials · 22 Ιουνίου, 2026 Τελευταίο άρθροΤο Evia Film Project επιστρέφειSpecials · 22 Ιουνίου, 2026
Τελευταίο άρθροΤο Evia Film Project επιστρέφειSpecials · 22 Ιουνίου, 2026 Τελευταίο άρθροΤο Evia Film Project επιστρέφειSpecials · 22 Ιουνίου, 2026


Η ζωή σε μια αρχαία πόλη δεν ήταν όπως τη φανταζόμαστε. Δύσκολα θα επιβίωνε εκεί ένας σύγχρονος άνθρωπος…

Καθώς διανύουμε τον μήνα του Pride, οι συζητήσεις γύρω από την queer εκπροσώπηση στα μέσα ενημέρωσης και στην ποπ κουλτούρα επιστρέφουν στο προσκήνιο. Ανάμεσα σε πολλά…





Εισαγωγή Η κολακεία αποτελεί ένα διαχρονικό φαινόμενο της ανθρώπινης συμπεριφοράς, στενά συνδεδεμένο με την εξουσία, την επιθυμία και την κοινωνική…





Θέλετε να διαβάσετε ένα μυθιστόρημα που θα σας ταξιδέψει στην καρδιά της…

Ο Oliver Sacks (1933–2015) υπήρξε ένας από τους γνωστότερους νευρολόγους παγκοσμίως. Ο…

Ο Derrida, όπως έχουμε ξαναναφέρει, θεωρούσε ότι πολλές φορές τα πάρεργα ενός…

Το Tell Me Lies έγινε ευρύτερα γνωστό στο κοινό μέσα από τη…

Αν έχεις βρεθεί ποτέ να χαζεύεις βιβλία χωρίς να μπορείς να αποφασίσεις…

Το βιβλίο «Κάτι χαμογελάει στις σκιές» του Ηλία Φουντούλη είναι μια συλλογή…

Οι Gemma είναι ένα πενταμελές συγκρότημα που δημιουργήθηκε το 2017 στο Ρέθυμνο Κρήτης. Αποτελείτε από…

Μακριά από τα συνηθισμένα τουριστικά μονοπάτια συναντά κανείς τις Αζόρες. Το αρχιπέλαγος κρύβει μερικά από τα πιο εντυπωσιακά τοπία της Ευρώπης. Τα εννέα νησιά του σχηματίζουν…

Το παρόν άρθρο, με τίτλο Παιδική κατάθλιψη: Είναι κρυμμένη στα μάτια; θα παρουσιάσει πρόσφατα ερευνητικά ευρήματα τα οποία δείχνουν ότι…





Αφιέρωμα στο «The New Look» κάνει ο Codd Kessler με την νέα…

Ο καιρός έχει αρχίσει σιγά σιγά επιτέλους να κρυώνει. Σε πολλές περιοχές,…

Πόσες είναι οι φορές που δε βρίσκουμε τίποτα να φορέσουμε και μας…

Καθώς μπήκε η wedding season, η αναζήτηση του τέλειου outfit είναι γεγονός.…

Είτε high είτε mid, τα πέδιλα θα είναι πάντα η λατρεία των…

Ανέκαθεν, τα λινά ρούχα θεωρούνταν “βασιλικά” υφάσματα, καθώς προορίζονταν κυρίως για τις…

Οι κοραλλιογενείς ύφαλοι αποτελούν αναπόσπαστο μέρος ενός θαλάσσιου οικοσυστήματος με το οποίο υπάρχει μια σχέση…

Τα ξημερώματα της 16ης Ιουλίου του 1945 στη έρημο του νέου Μεξικού, μια εντυπωσιακότατη λάμψη…

Δεν αγαπάμε όλοι με τον ίδιο τρόπο — και αυτό δεν είναι καθόλου πρόβλημα. Αντιθέτως είναι δύναμη. Η γλώσσα αγάπης που κουβαλάει ο καθένας μας, δεν…




Πρόσωπα, δημιουργοί και ιστορίες μέσα από τις πιο ενδιαφέρουσες συζητήσεις του MAXMAG.
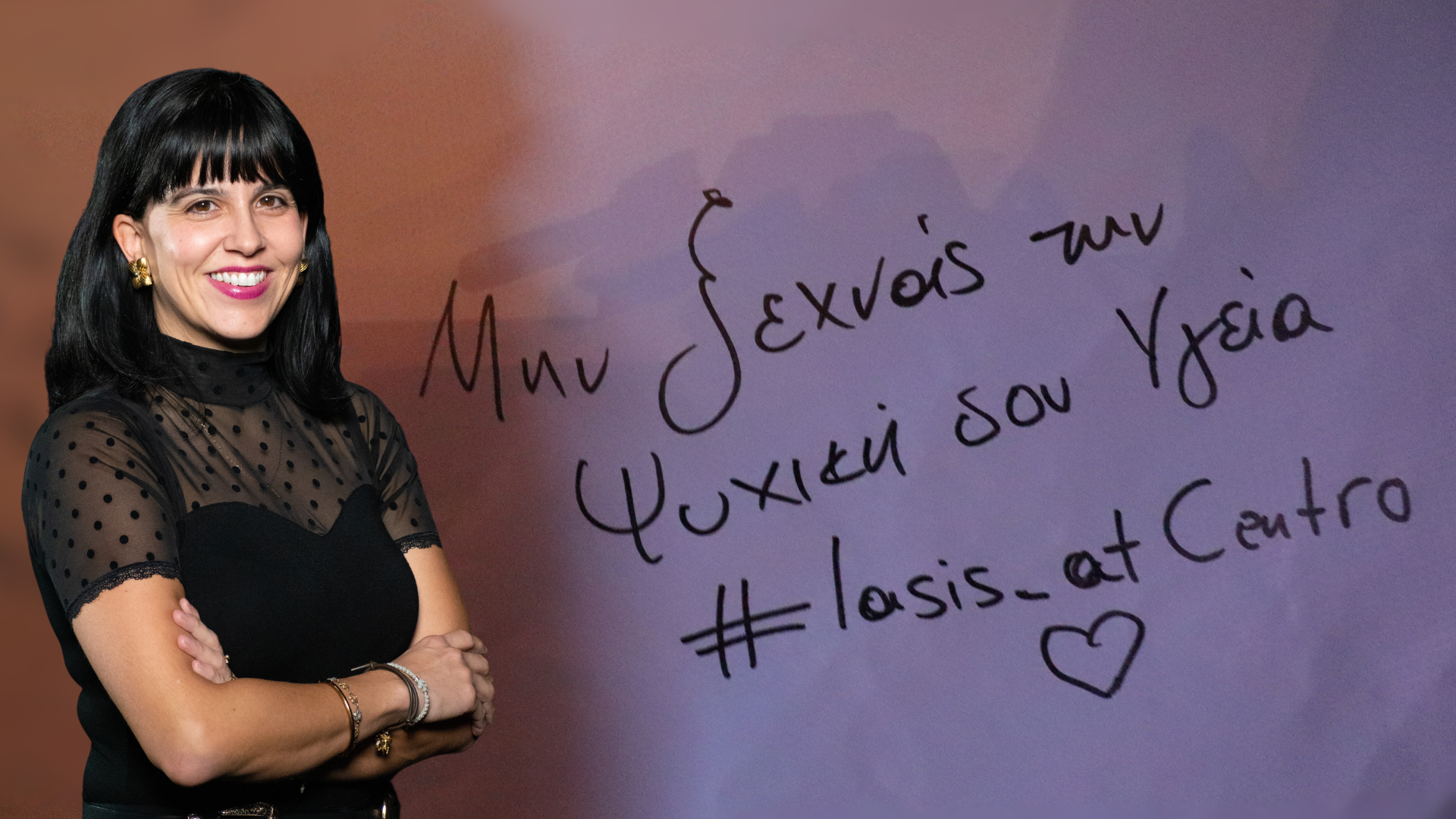
Θερμές ευχαριστίες στην κα Ιωάννα Αλπέρτη, Επιστημονικά Υπεύθυνη του Iasis at Centro, για την εμπεριστατωμένη συνέντευξή της. Μέσα από τις δράσεις της και τη δέσμευσή της στον τομέα…

Τι σημαίνει άραγε «να είμαστε εντάξει με τον εαυτό μας» ως γυναίκες; Και πως μπορούμε -η κάθε…

Ο Νίκος Μαντάς με τη γραφή του εκπλήσσει, προβάλλει το νόημα, το σημαντικό. Είναι ο σημερινός καλεσμένος…

Η Κλειώ Ιερωνυμάκη είναι μαζί στη στήλη των συνεντεύξεων για μια συζήτηση για το βιβλίο της “Ο…

Βρίσκεται ανάμεσα στις καλύτερες αθλήτριες στα 60 και 100 μέτρα με εμπόδια στην Ελλάδα. Η Σοφία Ιωσηφίδου,…
Μεγάλα θέματα, πρόσωπα και ιστορίες που αξίζουν περισσότερο χώρο και βαθύτερη ανάγνωση.
 Αφιερώματα
Αφιερώματα
Στις 4 Ιουνίου 1961, η παγκόσμια πολιτική σκηνή βρέθηκε σε ένα από τα πιο τεταμένα…
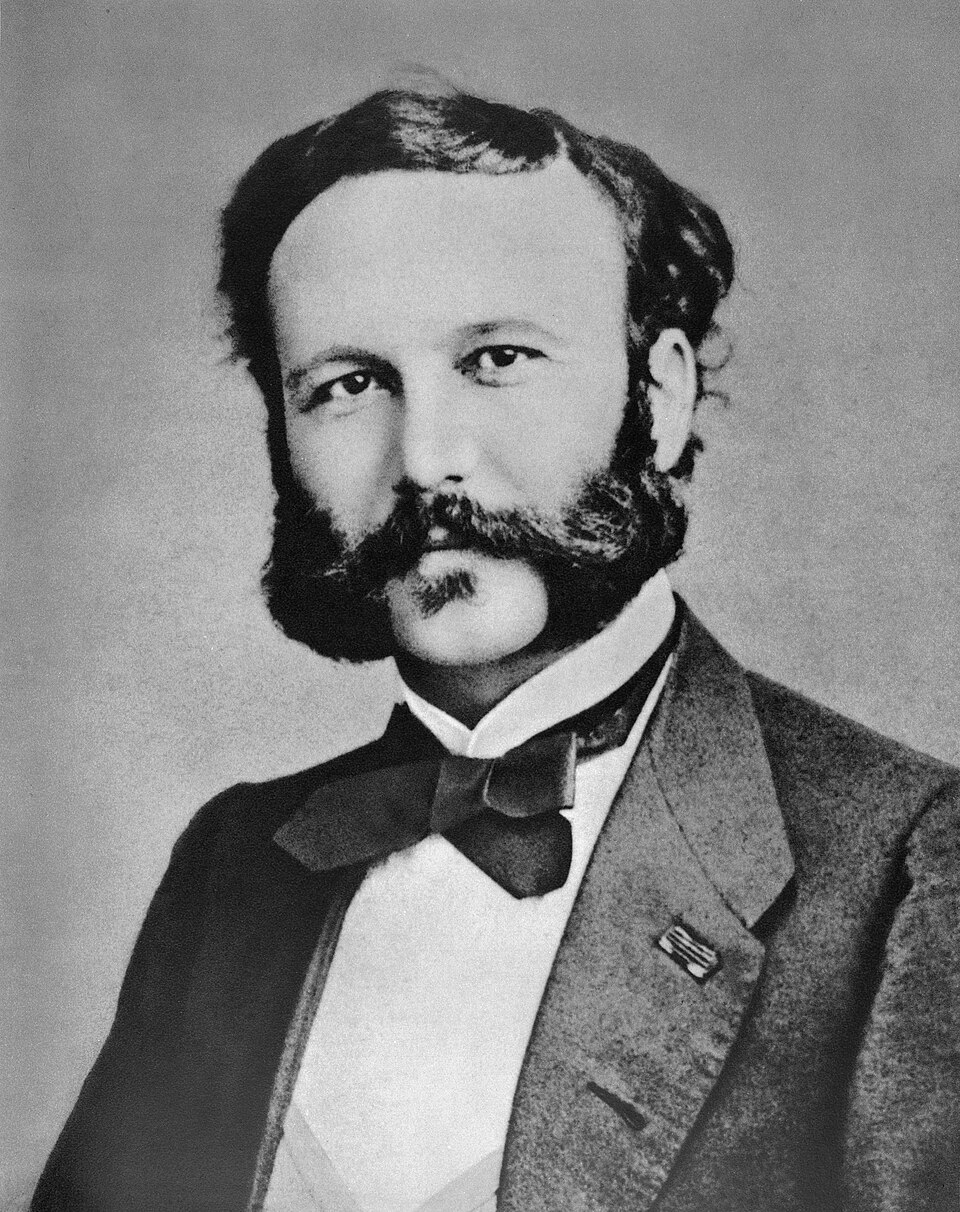 Αφιερώματα
Αφιερώματα
Ο Ερρίκος Ντυνάν (Jean Henri Dunant) ήταν ιδρυτής του Ερυθρού Σταυρού και ο πρώτος που…
Eurovision 1956: Η βραδιά που γεννήθηκε ένας ευρωπαϊκός θεσμός Σήμερα, η Eurovision αποτελεί ένα από τα μεγαλύτερα τηλεοπτικά και μουσικά…
 Αφιερώματα
Αφιερώματα
Το 1980 αποτέλεσε μια χρονιά-σταθμό για τη βιομηχανία των βιντεοπαιχνιδιών, καθώς τότε κυκλοφόρησε το θρυλικό…
 Αφιερώματα
Αφιερώματα
Η ευλογιά ήταν μολυσματική ασθένεια που προσέβαλλε αποκλειστικά τον άνθρωπο και την προκαλούσαν δύο στελέχη…
Κρυφές διαδρομές, χωριά και ελληνικά μέρη έξω από τους συνηθισμένους χάρτες.
Η πόλη σε πρώτο πλάνο: στέκια, έξοδοι, διαδρομές και μικρές διευθύνσεις που αξίζει να ανακαλύψεις.