
Πολιτισμός

Πολιτισμός
Ηρωίδα Αταλάντη: Η γυναίκα- σύμβολο

Πολιτισμός
Παράξενα έθιμα του ελληνικού καλοκαιριού
Το ελληνικό καλοκαίρι είναι γεμάτο από έθιμα που μπορεί να φαίνονται παράξενα και ιδιαίτερα σε έναν ξένο επισκέπτη,…




3285 άρθρα
Περισσότερα
Κινηματογράφος

Κινηματογράφος
Obsession | Το “αουτσάιντερ” του φετινού box-office
Obsession | 2026 | Διάρκεια 109′ | ★ IMDb (7.9/10), Letterboxd: (4.1/5), Rotten Tomatoes: 94% Από την ημέρα που κυκλοφόρησε στις αίθουσες, το Obsession δεν έχει…

Κινηματογράφος
Duel: Ένα αριστούργημα μινιμαλιστικού σασπένς

Κινηματογράφος
Ένας Νέος Γιος: Η δύναμη της παιδείας

Κινηματογράφος
The Innocents: Όταν το καλοκαίρι γίνεται εφιάλτης

Κινηματογράφος
Βοηθητικός ηθοποιός: Ο αόρατος επαγγελματίας
2365 άρθρα
Περισσότερα
Θέατρο

Θέατρο
Stand-Up Comedy: Η Εξέλιξη της Επιθεώρησης
Εισαγωγή Η ελληνική επιθεώρηση γεννιέται στις αρχές του 20ού αιώνα και εξελίσσεται σε ένα από τα σημαντικότερα λαϊκά θεάματα της…




3713 άρθρα
Περισσότερα
Βιβλίο

Βιβλίο
Για τις βουβές Φωνές του Αντόνιο Πόρτσια
Πλάι σ’ ό,τι συνήθως θεωρείται μοναδική μεγάλη λογοτεχνία, πλάι σε έργα ογκώδη,…

Βιβλίο
Château de Monte-Cristo, κραυγή αγωνίας για το «καταφύγιο» του Alexandre Dumas
Στους λόφους του Le Port-Marly, λίγα χιλιόμετρα δυτικά του Παρισιού, δεσπόζει ένα…

Βιβλίο
Out of Office, γιατί το διάβασμα στις διακοπές έχει άλλη γεύση;
Ας είμαστε ειλικρινείς. Πόσες φορές μέσα στον χειμώνα δεν κοιτάξατε εκείνο το…

Βιβλίο
“Everything Men Know About Women”, το βιβλίο που διαβάζεται σε… λίγα δευτερόλεπτα
Υπάρχουν βιβλία που αλλάζουν τον τρόπο που βλέπουμε τον κόσμο. Υπάρχουν βιβλία…

Βιβλίο
Η Βιβλιοθήκη που θα διαβάσουν οι δισέγγονοί μας
Στη Future Library τα βιβλία του σήμερα θα διαβαστούν από τους αναγνώστες…

Βιβλίο
Τα «ζωντανά» βιβλία του Harvard
Αν είσαι από εκείνους που φοβούνται μην τσαλακώσουν τις σελίδες, μην σπάσουν…
1724 άρθρα
Περισσότερα
Μουσική

Μουσική
Gemma: “Μέσα από την μουσική μπορούμε να βγούμε από τον λαβύρινθο της πραγματικότητας”
Οι Gemma είναι ένα πενταμελές συγκρότημα που δημιουργήθηκε το 2017 στο Ρέθυμνο Κρήτης. Αποτελείτε από…
1167 άρθρα
Περισσότερα
Ταξίδια

Ταξίδια
Μινόρκα:Το κρυμμένο διαμάντι των Βαλεαρίδων
9Μινόρκα Πηγή: content.r9cdn.net Μακριά από τους έντονους ρυθμούς της Ίμπιζα και την κοσμοπολίτικη ατμόσφαιρα της Μαγιόρκα, η Μινόρκα αποκαλύπτει μια πιο αυθεντική πλευρά της Μεσογείου. Το…




2076 άρθρα
Περισσότερα
Ψυχολογία
Ψυχολογία
Nαρκισσισμός και νοημοσύνη
Nαρκισσισμός και νοημοσύνη Τα άτομα με υψηλό ναρκισσισμό είναι ματαιόδοξα και μεγαλομανή. Έχουν ένα έντονο αίσθημα δικαιωματισμού, πιστεύοντας ότι αξίζουν…


Ψυχολογία
Ιδεοψυχαναγκαστική διαταραχή των σχέσεων

Ψυχολογία
Αλλάζει ο τύπος δεσμού;

29 άρθρα
Περισσότερα
Μόδα

Μόδα
Dior vs Chanel: Τα φαντάσματα πίσω από τους δύο θρύλους
Αφιέρωμα στο «The New Look» κάνει ο Codd Kessler με την νέα…

Μόδα
Τα πιο stylish και cozy looks για τα χιόνια
Ο καιρός έχει αρχίσει σιγά σιγά επιτέλους να κρυώνει. Σε πολλές περιοχές,…

Μόδα
Γκαρνταρόμπα: Τρόποι οργάνωσης για να φτιάχνεις εύκολα fashionable looks
Πόσες είναι οι φορές που δε βρίσκουμε τίποτα να φορέσουμε και μας…

Μόδα
Με αυτά τα outfits θα εντυπωσιάσεις εάν είσαι καλεσμένη σε γάμο
Καθώς μπήκε η wedding season, η αναζήτηση του τέλειου outfit είναι γεγονός.…

Μόδα
Τα πιο stylish πέδιλα για να φτιάξεις τα καλοκαιρινά looks
Είτε high είτε mid, τα πέδιλα θα είναι πάντα η λατρεία των…

Μόδα
Τα αγαπημένα μας καλοκαιρινά looks με τα λινά ρούχα
Ανέκαθεν, τα λινά ρούχα θεωρούνταν “βασιλικά” υφάσματα, καθώς προορίζονταν κυρίως για τις…
1321 άρθρα
Περισσότερα
Τεχνολογία & Επιστήμη

Επιστήμη
ΓΤΟ : Επιστήμη, Υγεία και Περιβάλλον
Οι άνθρωποι ήδη πριν από 10.000 – 12.000 χρόνια με στόχο την βελτίωση της καθημερινότητάς…
Επιστήμη
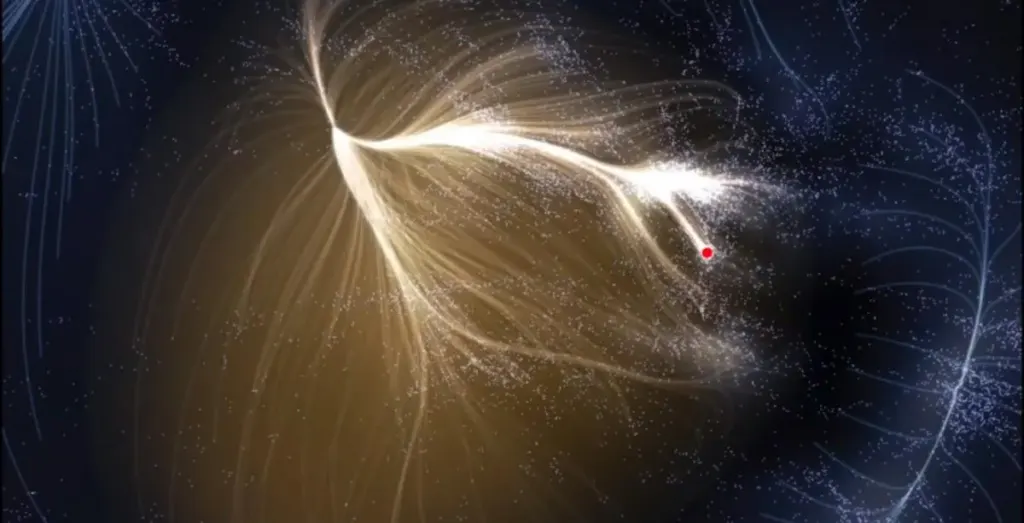
Ο Μεγάλος Ελκυστής: Η αόρατη δύναμη που τραβάει τον γαλαξία μας
Όταν κοιτάμε τον νυχτερινό ουρανό, τα άστρα μοιάζουν ακίνητα και γαλήνια. Στην πραγματικότητα, το σύμπαν…




1998 άρθρα
Περισσότερα
Αψυχολόγητα

Αψυχολόγητα
Love language: Η γλώσσα της αγάπης σου είναι η δύναμη σου
Δεν αγαπάμε όλοι με τον ίδιο τρόπο — και αυτό δεν είναι καθόλου πρόβλημα. Αντιθέτως είναι δύναμη. Η γλώσσα αγάπης που κουβαλάει ο καθένας μας, δεν…

Αψυχολόγητα
Οι Τρίτες μου δεν είναι πια ίδιες

Αψυχολόγητα
Μεταξύ «ιδανικών»

Αψυχολόγητα
Δεν φοβόμαστε την αγάπη, αλλά το κόστος της

Αψυχολόγητα
Κάθε πόνος είναι πέρασμα
MAXMAG Talks
Όλες οι συνεντεύξεις →
Συνεντεύξεις
Πρόσωπα, δημιουργοί και ιστορίες μέσα από τις πιο ενδιαφέρουσες συζητήσεις του MAXMAG.
MAXMAG Interview
“Η ψυχική υγεία ως γέφυρα για μια καλύτερη ζωή”: Συνέντευξη με την Ιωάννα Αλπέρτη
Θερμές ευχαριστίες στην κα Ιωάννα Αλπέρτη, Επιστημονικά Υπεύθυνη του Iasis at Centro, για την εμπεριστατωμένη συνέντευξή της. Μέσα από τις δράσεις της και τη δέσμευσή της στον τομέα…

Συνεντεύξεις
Μαρια Λεντζου: Η ομορφιά του να είσαι γυναίκα μέσω της Αυθεντικής Κίνησης
Τι σημαίνει άραγε «να είμαστε εντάξει με τον εαυτό μας» ως γυναίκες; Και πως μπορούμε -η κάθε…

Συνεντεύξεις
Νίκος Μαντάς: «Δεν μπορώ να φανταστώ έναν έρωτα να μοιράζεται»
Ο Νίκος Μαντάς με τη γραφή του εκπλήσσει, προβάλλει το νόημα, το σημαντικό. Είναι ο σημερινός καλεσμένος…

Συνεντεύξεις
Κλειώ Ιερωνυμάκη: «Έγραφα από μικρό παιδί»
Η Κλειώ Ιερωνυμάκη είναι μαζί στη στήλη των συνεντεύξεων για μια συζήτηση για το βιβλίο της “Ο…

Συνεντεύξεις
Σοφία Ιωσηφίδου: Στην Κορυφή των Εμποδίων στην Ελλάδα
Βρίσκεται ανάμεσα στις καλύτερες αθλήτριες στα 60 και 100 μέτρα με εμπόδια στην Ελλάδα. Η Σοφία Ιωσηφίδου,…
Variation 08
Δες όλα
Αφιερώματα
Μεγάλα θέματα, πρόσωπα και ιστορίες που αξίζουν περισσότερο χώρο και βαθύτερη ανάγνωση.
 Αφιερώματα
Αφιερώματα
Eurovision 1956: Η βραδιά που γεννήθηκε ένας ευρωπαϊκός θρύλος
Eurovision 1956: Η βραδιά που γεννήθηκε ένας ευρωπαϊκός θεσμός Σήμερα, η Eurovision αποτελεί ένα από…
 Αφιερώματα
Αφιερώματα
4 Ιουνίου 1961: Η κρίση του Βερολίνου
Στις 4 Ιουνίου 1961, η παγκόσμια πολιτική σκηνή βρέθηκε σε ένα από τα πιο τεταμένα…
Irena Sendler: Η γυναίκα που έσωσε 2.500 παιδιά από το Ολοκαύτωμα
Μια γυναίκα που στάθηκε απέναντι στην ιστορία Στη σκοτεινότερη περίοδο της ευρωπαϊκής ιστορίας, όταν ο φόβος και η βαρβαρότητα κυριαρχούσαν,…

Hidden Greece
Περισσότερα
Άγνωστη Ελλάδα
Κρυφές διαδρομές, χωριά και ελληνικά μέρη έξω από τους συνηθισμένους χάρτες.





Variation 12
Όλα στο City Guide
City Guide
Η πόλη σε πρώτο πλάνο: στέκια, έξοδοι, διαδρομές και μικρές διευθύνσεις που αξίζει να ανακαλύψεις.





